|
|
 发表于 2014-7-14 20:05:25
|
显示全部楼层
发表于 2014-7-14 20:05:25
|
显示全部楼层
GO Fight Against Malaria update: promising early findings for malaria & drug-resistant tuberculosis
By: Dr. Alexander L. Perryman
14 七月 2014
摘要
Dr. Alexander Perryman describes the analysis and initial findings from the first phase of GO Fight Against Malaria, which include the discovery of several promising hits against key drug targets for treating both malaria and drug-resistant strains of tuberculosis. They are conducting further analysis and experimentation on the massive amount of data generated by World Community Grid volunteers.
Dear fellow volunteers of World Community Grid,
In under two years, World Community Grid volunteers performed the world's largest docking project, carrying out over 1 billion calculations to help us identify chemical compounds to advance the treatment of increasingly drug-resistant strains of malaria and other diseases - a process that would have taken over a hundred years on the type of computer clusters currently available at most universities.
Since we completed GO Fight Against Malaria (GFAM) calculations on World Community Grid a year ago, we've been analyzing the generated data. Although that process will continue for some time still, early analysis has revealed several promising findings.
First, we identified the first "small molecule" inhibitor (i.e., drug-like compound) to block the activity of a particular malaria enzyme involved in infection, the first step in developing a potential treatment or prevention aimed at this malaria drug target.
Also, a subset of your calculations was conducted against a drug target for malaria which shares a similar atomic structure to a Mycobacterium tuberculosis enzyme. With extensively drug-resistant strains of tuberculosis on the rise, there is a pressing need to identify more effective treatments. We therefore included this particular tuberculosis drug target in our GFAM experiments. In doing so, we have identified several chemical compounds as potential inhibitors of this enzyme and have confirmed these results with initial laboratory tests. A very impressive number of the promising chemical compounds identified through the virtual screenings you computed on World Community Grid have gone on to perform well in additional lab testing: 20% were "hits", vs. less than 1% on average for other experimental ("wet lab") high-throughput tuberculosis experiments.
We are now designing and synthesizing new derivatives of these inhibitors to further refine them as viable drug candidates. Read on for more details about this early analysis work, and we'll be able to share more information once we publish our findings. In the meantime, I want to thank GFAM volunteers for allowing us to advance this important and often neglected area of research.
Largest set of computational docking experiments ever performed
GFAM was launched on IBM's World Community Grid on November 16, 2011. Malaria is one of the three deadliest infectious diseases on Earth (the other two are HIV and tuberculosis). Plasmodium falciparum (Pfal, or Pf), the species that causes the worst form of malaria, kills more people than any other parasite on the planet. Over 200,000,000 clinical cases of malaria occur each year, and over one million people are killed by malaria every year. Over three billion people (almost half of all humans) are at risk of becoming infected with malaria, and every 30 seconds, another child dies of malaria.
GFAM ran on World Community Grid for 19 months, during which the tremendous computational power provided by World Community Grid volunteers like you helped us generate massive data sets against 22 different types of drug targets, to seed the discovery of new drugs to treat malaria. We performed "docking calculations," which explore how well different "small molecules" (pieces of drug-like compounds) are able to bind and potentially block the activity of critical pieces of the molecular machinery that the pathogens use to survive, replicate, and spread throughout humanity. Docking calculations use flexible models of these small molecules to explore the energetic landscape of atomic-scale models (on the scale of 0.0000000001 meters) of proteins that perform critical functions for the parasite's lifecycle and infection process. These calculations predict how tightly a compound might bind to the target (that is, how potent it might be), where the compound probably prefers to bind, and what specific types of interactions might be formed between the compound and the drug target. One docking calculation refers to the process of docking a flexible model of a single compound against one particular version of one target. In this first phase of GFAM, World Community Grid volunteers performed 1.16 billion different docking calculations that explored the potential activity of 5.6 million different compounds against drug targets from malaria (and against some targets for treating drug-resistant tuberculosis, Methicillin-Resistant Staphylococcus aureus (MRSA), filariasis, and bubonic plague, when the targets from those other pathogens had structural similarity to the targets from malaria). With the computing power that you generously donated, GFAM was the first project to ever perform a billion different docking calculations. Performing this many calculations could have taken over a hundred years on the type of computer clusters currently available at most universities. We could not have accomplished this feat without your help. We are also grateful for the $50,000 in seed funding provided by the IBM International Foundation, from part of the prize money that IBM's computer Watson won on Jeopardy!™. Thus far, that seed money has been the only funding that the project has received, but we are currently writing grants that focus on analyzing and extending the GFAM data.
Finding a "hit" that inhibits a critical protein involved in malaria infections
"Hits" are compounds that have some inhibitory effect on the biological activity of one of these drug targets. But finding a hit is only the beginning of the process (a complicated process that can take several years to a couple of decades to complete). Scientists from around the world called "medicinal chemists" can then work with structure-based computational chemists like us to try to increase the potency and decrease the potential toxic side effects of these compounds, which involves processes called "hit-to-lead development" and then "lead optimization". "Leads" are larger, more structurally complex, potential drug candidates that generally display nanomolar potency (that is, they are around 1,000 times more potent than "hits", which means that only a tiny amount of a leading compound is required to affect the activity of the target). In collaboration with Professor Mike Blackman's lab in the Division of Parasitology at the Medical Research Council's (or "MRC's") National Institute for Medical Research (or "NIMR"), in London, UK, and with InhibOx, Ltd, we searched for the first small molecule inhibitors of the potential drug target "PfSUB1" (see target class #6 onhttp://gofightagainstmalaria.scripps.edu/index.php/how-we-will-discover-potential-malaria-drugs). When the Blackman lab solved the first crystal structure of PfSUB1 (that is, the atomically-detailed, 3-D map of where all of its atoms are), they shared that unpublished structure with us, which allowed us to perform virtual screens against PfSUB1. These virtual screens are searching for "small molecule" inhibitors (that is, compounds with some similarity to pieces of known drugs) that can block the activity of this malarial enzyme. When malaria parasites replicate themselves inside a red blood cell, the "daughter" parasites eventually rupture the infected host cell, which allows the new parasites to escape and then invade and infect other red blood cells. The subtilisin-like serine proteases from Plasmodium falciparum (also known as PfSUB1) are involved in this ability of the malaria parasites to escape (or "egress") an infected red blood cell. The Blackman lab has shown that the PfSUB1 enzyme has an additional role in "priming" the merozoite stage of the parasite prior to its invasion of red blood cells (in other words, it is involved in processing certain other malarial proteins in order to prepare and activate them, so that the parasite can invade our blood cells). Thus, PfSUB1 is involved in both the egress and the infection process. In the results of GFAM Experiment 27, we discovered the first small molecule inhibitor of PfSUB1 ever identified, and it displayed a proper "dose-response curve" (that is, at higher concentrations of the inhibitor, it shuts down the activity of PfSUB1 more and more effectively), which indicates that it is likely a "specific" inhibitor, instead of a non-specific compound that randomly happens to impede activity a bit for many different types of proteins (but this will have be tested against other types of proteins to know for sure). This compound, nicknamed "GF13", is a fairly weak inhibitor: at a 200 micromolar concentration, it blocks activity of PfSUB1 50%. Strong hits will block 50% of the target's activity in the 1 to 50 micromolar range (the smaller the # of micromoles per liter that are needed to shut down activity, the more potent a compound is).
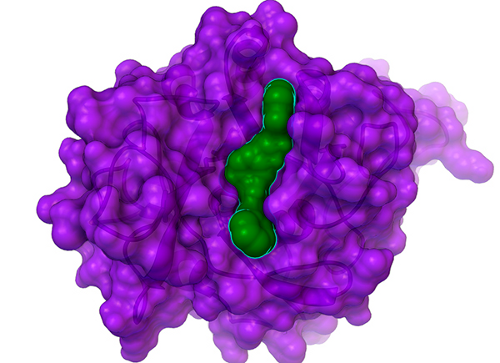
Although GF13 (shown as the green surface with a cyan outline that is bound in the cleft in the center of PfSUB1, whose surface is shown in purple above) is a weak inhibitor, it is still a novel and significant hit: it provides a foundation on which we can build, and it could help us find more potent inhibitors of this potential drug target for malaria. After we write a couple papers on the GFAM results against the tuberculosis target InhA (discussed below), which could help us get some grant funding to enable additional analyses of GFAM data, we will write a paper on these results against PfSUB1. We then hope to extend this collaboration with Professor Mike Blackman's lab on this important malaria target (if we can obtain a grant to enable that extension).
GFAM experiment leads to the discovery of new hits against a key drug target for tuberculosis.
As mentioned earlier, when a drug target from Plasmodium falciparum had structural similarity with a target from another pathogen, we docked 5.6 million compounds against the targets from both pathogens. A potential drug target for malaria called "PfENR" (for Plasmodium falciparum enoyl acyl-carrier-protein reductase) has a similar atomic structure to the well-validated drug target for treating tuberculosis called "InhA". Consequently, we included theMycobacterium tuberculosis (Mtb) enzyme InhA in our GFAM experiments against PfENR. Few pharmaceutical companies perform antibiotic (i.e., antibacterial) research anymore, which means that it is up to scientists at universities and non-profit institutes to fill that research gap. If the scientific community cannot create new ways to defeat these superbugs, then the medical community will not have the capacity to treat these drug-resistant infections that keep occurring with increasing severity, frequency, and distribution. The situation is dire. Mtb infects 8.3 - 9 million people each year, and tuberculosis kills 1.4 million people/year. A few decades ago, multi-drug-resistant tuberculosis (MDR-TB) was not a serious problem. There are now a half million new cases of MDR-TB per year. Extensively drug-resistant TB (XDR-TB) has now been found in over 92 different countries, including the U.S.A. And "nosocomial" (i.e., hospital-acquired) XDR-TB is now a growing problem. Totally-drug-resistant TB (TDR-TB) has appeared in several countries and will continue to spread. No drugs exist to treat TDR-TB.
Professor Peter J. Tonge is the first person we encountered who was willing and able to test some of the predictions from GFAM, even though it is an unfunded project. Professor Tonge is the Director of Infectious Disease Research at the Institute for Chemical Biology and Drug Discovery at Stony Brook University in NY. He has done some pioneering research against MtbInhA and is a leading expert in the battle against XDR-TB. He offered to experimentally assess the potency of the candidate compounds we discovered in the docking calculations on GFAM that we performed against InhA. InhA (which is also called FabI) is part of a unique metabolic pathway (that is, it's an enzyme that is part of a metabolic pathway that is not present in humans), which should hopefully decrease the toxic side effects of InhA inhibitors. Specifically, ENR/InhA is part of a Fatty Acid Synthesis pathway (called "FAS II") that human cells do not have. One of the main drugs used to treat tuberculosis is called isoniazid (or "INH"), and it kills that deadly bacteria by shutting down the activity of InhA (and perhaps by also shutting down the activity of other target proteins in Mtb, as well). But drug-resistant mutants against which isoniazid loses its effectiveness keep evolving and spreading, which is why we are searching for new inhibitors of InhA that are able to counteract the main mechanism of drug resistance that allows Mtb to evade treatment with INH. Some inhibitors of InhA also block the activity of the PfENR target from malaria. By advancing the research against InhA, we might be able to simultaneously help advance the research against both totally drug-resistant tuberculosis and against multi-drug-resistant malaria.
In the results of GFAM Experiment 5, which screened the National Cancer Institute's library of compounds (that we can order for free from the NCI's Developmental Therapeutics Program), we identified 19 candidate compounds as potential inhibitors of Mtb InhA. These 19 NCI compounds were then experimentally tested in "wet lab" experiments (in test tubes and Petri dishes) by Weixuan Yu in Professor Peter Tonge's lab. Of the 16 soluble compounds, 8 candidates (at a 100 micromolar concentration) shut down InhA activity by ~ 30% or more. The most potent inhibitor we discovered displayed an IC50 value of ~ 40 micromolar (which means that when the compound is present at a 40 micromolar concentration, it inhibits InhA activity by 50%). Additional kinetic experiments were then performed on the best hits from this experiment, and the two most potent inhibitors displayed Ki values of 54 and 58 micromolar.
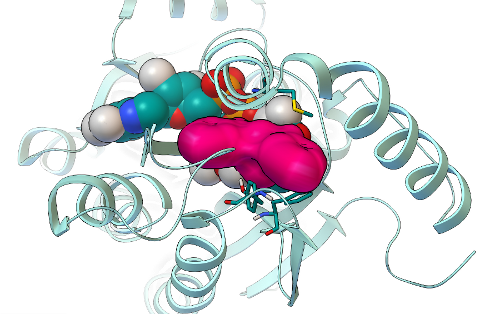
The binding mode predicted by AutoDock Vina for the most potent hit we discovered in GFAM experiment 5 is shown as a magenta surface, while the InhA target is shown in cyan as a ribbon above. The NAD cofactor of InhA is shown as cyan spheres. Finding new, low micromolar inhibitors of InhA is a significant achievement (and so is having a hit rate of 8/19 candidates from a virtual screen), but we will still need to optimize these compounds and make them at least a thousand times more potent before they become a drug-like candidate called a "lead". These virtual screens on GFAM (and, thus, the new inhibitors we discovered) were designed to target one of the main mechanisms that Mycobacterium tuberculosis has evolved in order to resist the effects of drug treatment with isoniazid. We are now about half-way through the process of writing a paper on these exciting new results. I hope to finish the first draft within the next month or so, and then I'll send it around to all of the co-authors to get their input and suggested revisions. After that manuscript completes the normal peer-review process, we will share the published version with all of you.
Discovery of additional new hits against a target for drug-resistant tuberculosis
The third GFAM experiment we analyzed led to the discovery of additional new hits against InhA, a key drug target for treating Mycobacterium tuberculosis, and could help seed the discovery of new drugs against extensively drug-resistant and totally drug-resistant tuberculosis.
In November of 2013, I joined Associate Professor Joel S. Freundlich's chemical biology and medicinal chemistry lab at the Rutgers University New Jersey Medical School, in Newark, NJ. Instead of having to search for collaborators in other labs to experimentally test my predictions, and then waiting many months to years to find out whether the compounds actually work against the drug targets and then against the full pathogenic organism, I can now just ask my co-workers in the Freundlich lab to help test the computational predictions. The enzyme activity assays, anti-bacterial activity assays against whole-cell Mtb cultures, metabolomics studies to investigate the different targets that are affected, synthesis of new derivatives, and medicinal chemistry-guided optimization research are all performed within the Freundlich lab. I provide the computational chemistry core that can help analyze and guide some of these efforts. I've even started learning how to do some of these "wet lab" aspects myself, to expedite the assessment of my computational predictions. In the Freundlich lab, some of our projects againstMtb involve analyzing and extending the subset of GFAM data that involve targets that can advance the treatment of XDR-TB and TDR-TB.
In GFAM experiment 9, which involved docking the Asinex library of over 500,000 compounds against PfENR and Mtb InhA, we discovered additional new inhibitors of InhA that have very novel structures, as compared to the known InhA inhibitors. In collaboration with Dr. Sean Ekins of Collaborations in Chemistry (Fuquay-Varina, NC), this project also involves the creation, testing, and development of new types of computational workflows that can filter and prioritize the docking results, in order to increase efficiency and enhance the probability of finding active, potent, non-toxic compounds. We used an innovative new combination of docking-based filters, other types of filters (which we cannot discuss until after these results are published), and visual inspection to narrow down the docking results for those 500,000 compounds and identify 20 promising candidate compounds. These 20 Asinex compounds were then tested in enzyme inhibition assays against InhA by Xin Wang, a graduate student who recently joined the Freundlich lab. Since Professor Freundlich was a medicinal chemist in the pharmaceutical industry for over a decade, he applied a more stringent bar to define what we would consider as hits: a "hit" had to inhibit InhA activity by at least 50% when the compound was present at a 50 micromolar concentration. Using this more stringent criteria, 5 of the 20 candidates are novel hits against InhA. The most potent new InhA inhibitor we discovered has an IC50 of 19 micromolar (at a concentration of 19 micromoles per liter, it inhibits InhA activity 50%).
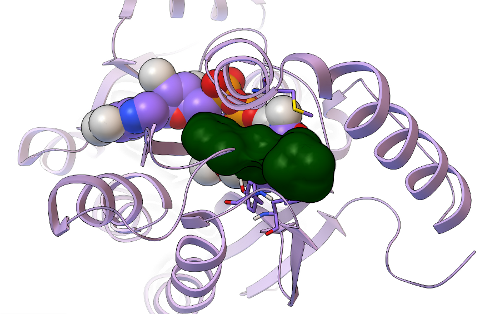
This new hit's predicted binding mode against InhA is shown as a dark green surface, with the InhA target shown in light purple as a ribbon above. Having a 5/20, or 25%, hit rate against an enzyme from the results of a virtual screen was a great success, as was discovering another novel InhA inhibitor that was more than twice as potent as the one we found in our previous GFAM experiment against InhA. The 5 hits we discovered against InhA were then further tested for their activity against whole-cell Mycobacterium tuberculosiscultures by Dr. Mi-Sun Koo in the Freundlich lab. These "MIC99" assays (for the Microbial Inhibition Constant, or the amount of compound needed to inhibit the growth of 99% of the bacterial cells) take over a week to perform againstMtb, since Mtb grows very slowly. Because Mtb is an air-borne bacteria, these MIC assays have to be done in special "BSL3" labs (the second highest form of Biosafety Level), using the full-body "bunny suits" and respirators. Since Dr. Koo went on vacation before the assays were done, the final measurement was taken by Dr. Pradeep Kumar, of Professor David Alland's lab at the Rutgers University-NJ Medical School. Of the 5 InhA inhibitors we discovered, one of these compounds displayed potency against whole-cell cultures of the bacteria that cause tuberculosis, with an MIC99 of ~ 2.4 micrograms per milliliter.
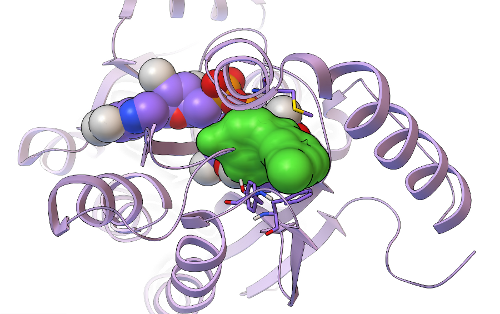
Its predicted binding mode is shown as a light green surface, with the InhA target displayed in light purple as a ribbon. Traditional, experimental high-throughput screens against whole-cell Mtb tend to display hit rates less than 1%, but 1/5 (i.e., 20%) of the compounds we tested were active against whole-cell Mtb cultures. Professor Joel Freundlich, Dr. Shao-Gang Li, Dr. Steve Paget, and I have already designed new derivatives of the top InhA inhibitor and the top MIC hit, to test their Structure-Activity-Relationships (to figure out how changing and adding different chemical groups to these molecules increases or decreases their activity) and to try to increase their potency. These derivatives are currently being synthesized by Shao-Gang and Steve, as part of the "hit-to-lead development" process mentioned previously. After these derivatives have been made, tested in InhA activity assays, and tested in Mtbgrowth assays, we will write a paper on these exciting new results. Professor Freundlich, Dr. Sean Ekins, and I have already written and submitted two different grants that discuss our new approach and propose applying it to the other 5 million compounds that were docked against InhA as part of the GFAM project, and we plan to write a couple more grants that involve the Mtb subset of GFAM data. We'll let you know how it goes.
Personnel update and a request for action
Last year, the "sequester cuts" to the NIH eliminated the funding for my position in Professor Olson's lab at TSRI. Don't be sad for me - please contact your members of Congress and ask them to restore funding to the NIH! The inflation in scientific equipment and reagents rises faster than normal inflation, but the NIH budget has still not yet been restored to even the pre-sequester level. It's not even close to the level of funding it should have, considering scientific inflation, the growing problems that the medical community faces, and the fact that pharmaceutical companies keep cutting their research budgets year after year (which puts the academic scientific community under pressure to fill this research gap, or else progress against diseases will not occur rapidly enough). In addition, every single dollar from the NIH and NSF generates approximately $2 in economic output, and NIH funding has led to the creation of nearly 430,000 quality jobs. Luckily, I was able to find a great new position in Professor Joel Freundlich's lab at the Rutgers University-New Jersey Medical School, where I get to continue my fight against drug-resistant infectious diseases. But I know many scientists who are still not able to find a job.
Thank you all very much for donating your computer power to World Community Grid!!! We would not have been able to generate this mountain of data for malaria and tuberculosis research without your generous help. The massive number of virtual experiments you performed provides us with enough data to continue our research for years to come. Please be patient, and keep crunching! Many other projects still need your help.
Sincerely,
Dr. Alex L. Perryman
大意:
GFAM项目更新:发现潜在的治疗疟疾和耐药性肺结核药物靶点
GFAM已经在一年前完成了计算,现在我们在分析结果数据,以寻找治疗抗药性疟疾(以及其他疾病)的药物。
首先,我们找到了一个小分子抑制剂,可用于抑制疟疾在感染过程中用到的一种酶。
同时,我们还针对另外一种疟疾和肺结核杆菌通用的酶进行了计算。我们找到了几种潜在药物,并在试验中进行了初步测试。结果很喜人,抑制率高达20%,而其他药物只有1%。现在我们正在优化设计和合成这些抑制剂,以便将来能用于临床治疗。
GFAM开始于2011年11月16日,地球上最致命三大传染病疾病是艾滋、肺结核和疟疾。恶性疟原虫是导致疟疾的元凶,其中每年死于疟原虫的人数远超过其他寄生虫。每年大约有2亿人次临床感染记录,其中有超过1百万人死于疟疾。全球大约有30亿人口都在疟疾高危区。每半分钟就有一个儿童死于疟疾。
GFAM在WCG上运行了19个月。它利用结合能计算,来筛选能抑制病菌生长、复制、感染的分子药物。志愿者们完成了11.6亿次结合能计算,找到了560万种可能用于抑制疟疾(有些还能用于耐药性肺结核、抗药性的金黄葡萄状球菌MRSA、丝虫病、淋巴腺鼠疫,因为他们的结构与疟疾酶类似)的分子。感谢志愿者们的无私奉献,也感谢IBM提供的5万美元赞助(来自于沃森在Jeopardy节目中赢得的奖金)。
寻找疟疾传染蛋白酶的靶点
寻找靶点是药物研发的第一步,找到后需要对药物进行优化,及去毒性处理(整个药物研发过程很漫长,大概需要几年到数十年)。直到找到真正有效的药物(最终的临床药物要比初筛的药物更有效1千倍以上才行),也就是只需要很小的剂量就能起效。我们和Mike Blackman教授合作,发现了PfSUB1目标蛋白酶,他把未公开的蛋白质3D结构数据给了我们,让我们进行药物筛选。疟原虫在感染红细胞后会在红细胞内进行疯狂繁殖,然后胀破红细胞,继续感染其他红细胞。PfSUB1蛋白酶在入侵和胀破红细胞膜的过程中起了关键作用。在GFAM的27号模拟实验中,我们发现了一直潜在抑制剂GF13。不过它的效用还比较差,200微摩尔浓度的GF13能使PfSUB1活性下降50%。有效药物,应该能在1-50微摩尔的浓度下使PfSUB1活性下降50%。
虽然GF13的效用比较差,但是它至少打了个好基础。现在我们在写肺结核目标蛋白酶InhA的论文,等写完了,应该可以拉不少赞助,然后我们就有钱继续疟疾的研究了。
GFAM实验发现新的肺结核药物靶点
如前所述, 疟原虫的靶蛋白酶PfENR和肺结核的靶蛋白酶InhA在结构上有点接近,所以我们把可能有效的560万种分子,针对两种靶蛋白酶都进行了计算。当前很少有制药公司会研究靶向抗菌药,所以只有大学学者和非营利性机构来干这活儿。要知道每年有830-900万人感染肺结核,其中有140万死于该疾病。几十年前,耐药性肺结核杆菌还不是个事儿,但是现在每年大约有50万人(92个国家)感染耐药性肺结核。更可怕的是,现在出现了完全耐药性肺结核。目前没有任何药物对它管用。
Tonge教授,是肺结核方面的专家,也是我们遇到的一个愿意自费进行研究的学者(译注:向这位仁兄致敬)。InhA是一种肺结核专用的新陈代谢酶,这意味着针对它的抑制剂,靶向性更好,毒副作用更低。当前治疗肺结核的主要药物是异烟肼(INH),它通过抑制InhA(以及其他蛋白酶)的活性来杀死病菌。但是现在病菌的变异使它对INH产生了抗药性,所以我们急需研发新的药物。而且对肺结核InhA的研究和对疟疾的研究都是相辅相成,互相促进的。
在GFAM实验5中,我们对NCI数据库中的分子进行了筛选。发现了19种候选分子,随后在实验室进行了测试。在16种可溶性分子中,8种在100微摩尔浓度下使InhA活性下降了30%。最有效的分子,在40微摩尔浓度下使InhA活性下降50%,亚军是54微摩尔,季军是58微摩尔。
由AutoDock Vina生成的GFAM实验5图中,靶点是红紫色的表面,而InhA酶是上面的青绿色的带状物,InhA的NAD辅助因子是青绿色的球面。此次发现非常振奋人心,因为它可以以极低的浓度产生效用。当然真正用于临床的药物要比这有效上千倍才行。所以我们还要继续优化。目前我们正在写论文,已经写了一半了,预计下个月完成草稿。待完成同行评审发表后,我们会公开论文。
发现新的抗药性肺结核靶点
在GFAM的实验3中,我们发现了新的InhA靶点。在GFAM的实验9中,我们对Asinex库中的50万个分子进行了筛选,发现与现有的抑制剂结构完全不同的新抑制剂。最终我们找到了20种候选分子并在实验室进行了测试。测试后,发现有5种分子在50微摩尔浓度下可以抑制50%的InhA,其中最好的可以低至19微摩尔。
新靶点表面为暗绿色,InhA酶是淡紫色带状物。老靶点的有效率高达20%,但是新靶点比它多一倍。我们对5个候选药物进行了肺结核病毒全细胞MIC99标准测试(即对病毒细胞生长抑制率达到99%),最终我们发现,其中一种分子,能在2.4微摩尔浓度下达到MIC99标准。我们现在针对这个分子还在不断的进行优化和调整。待有新的结果后我们会通知大家的。
个人吐槽
去年NIH大幅缩减了财政预算,这导致医学研究经费日趋紧张,我也因此失业(不过还好现在找到了新工作),如果你有‘舅舅’在米国国会,请让他们给NIH增加预算吧。现在制药企业在逐年缩减科研支出,国家方面也在减。要知道NIH和NSF的每一美元投入最终都能得到2美元的产出。而且NIH还能产生43万个相关工作岗位。要知道我还有很多同事根本找不到工作呢。
最后由衷的感谢大家的付出。
译注:翻译了一天终于翻译完了,休息一下,累屎我了。
|
评分
-
查看全部评分
|



 发表于 2014-10-4 21:54:26
发表于 2014-10-4 21:54:26
 Bronze - earned for 1 member recruited
Bronze - earned for 1 member recruited Silver - earned for 5 members recruited
Silver - earned for 5 members recruited Gold - earned for 10 members recruited
Gold - earned for 10 members recruited Ruby - earned for 25 members recruited
Ruby - earned for 25 members recruited Emerald - earned for 25 members recruitedWe hope that some enterprising members will force us to create a diamond-level badge as well!
Emerald - earned for 25 members recruitedWe hope that some enterprising members will force us to create a diamond-level badge as well!
